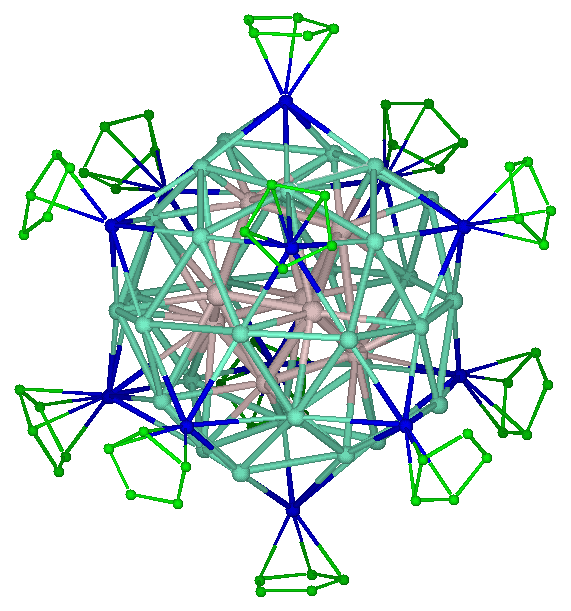
 Metalloid cluster: Al50Cp12
Metalloid cluster: Al50Cp12
Master Research Project 2: Theoretical Study of Nanometric Scale Metalloid Clusters
2006, 6 months : Université de Rennes 1 – Laboratoire de Chimie Théorique Inorganique
Supervisor: Dr J.-Y. Saillard
Metalloid clusters, or elementoid clusters, are formed by a small cluster of metal atoms surrounded, and protected by a shell of ligands. Some famous metalloid clusters have core formed by tens of metal atoms, such as Ga84[N(SiMe3)2]20 or Al77{N(SiMe3)2}20 [1]. For fundamental research, they are interesting subject of studies. Indeed, on a structural point of view they tend to adopt a metal structure in the core while the outer shells favor molecular structures. The modelling of these structures were performed using the Amsterdam Density Functional package in order to study the intimate relationship between the number of electrons and the local structure of the metal atoms. Metalloid clusters can be considered as formed by several shell of atoms. Due to their sizes, clusters are spherical and generally the structure adopted by a shell of metal atoms has a high symmetry. Depending of the number of metal atoms, the inner shell is square based antiprism (Al50), atom-centered dodecahedron (Al69) or atom-centered icosahedron (Al77). The core shell of metal atoms being surrounded by metal atoms tends to adopt a close packing structure that are very usual for metals (as face-centered cubic or hexagonal close packing structures). Accordingly the high symmetries structures described above are distorted towards hexagonal packing, and this distortion propagates to the outer shell.
Reference:
[1] Formation, structure and bonding of metalloid Al and Ga clusters. A challenge for chemical efforts in nanosciences, H. Schnöckel, Dalton Transactions, 4344-4362 (2008). DOI: 10.1039/B718784J
2006, 6 months : Université de Rennes 1 – Laboratoire de Chimie Théorique Inorganique
Supervisor: Dr J.-Y. Saillard
Metalloid clusters, or elementoid clusters, are formed by a small cluster of metal atoms surrounded, and protected by a shell of ligands. Some famous metalloid clusters have core formed by tens of metal atoms, such as Ga84[N(SiMe3)2]20 or Al77{N(SiMe3)2}20 [1]. For fundamental research, they are interesting subject of studies. Indeed, on a structural point of view they tend to adopt a metal structure in the core while the outer shells favor molecular structures. The modelling of these structures were performed using the Amsterdam Density Functional package in order to study the intimate relationship between the number of electrons and the local structure of the metal atoms. Metalloid clusters can be considered as formed by several shell of atoms. Due to their sizes, clusters are spherical and generally the structure adopted by a shell of metal atoms has a high symmetry. Depending of the number of metal atoms, the inner shell is square based antiprism (Al50), atom-centered dodecahedron (Al69) or atom-centered icosahedron (Al77). The core shell of metal atoms being surrounded by metal atoms tends to adopt a close packing structure that are very usual for metals (as face-centered cubic or hexagonal close packing structures). Accordingly the high symmetries structures described above are distorted towards hexagonal packing, and this distortion propagates to the outer shell.
Reference:
[1] Formation, structure and bonding of metalloid Al and Ga clusters. A challenge for chemical efforts in nanosciences, H. Schnöckel, Dalton Transactions, 4344-4362 (2008). DOI: 10.1039/B718784J
Master Research Project 1: Test of a Reverse Non-Equilibrium Dynamic Molecular Method for the Calculation of Thermal Conductivities of Solids
2005, 4 months : National Research Council Canada
Supervisor: Dr D. D. Klug
The thermal conductivity of a material is quantity that characterizes the heat transfer of a material. It is the result of two contributions, the electronic conductivity and the phonon conductivity. The method tested here is used to evaluate the phonon conductivity of a material prior its implementation to the density functional theory code Demon [1].
To compute the thermal conductivity of a system from molecular dynamic simulations, several methods are possible:
2005, 4 months : National Research Council Canada
Supervisor: Dr D. D. Klug
The thermal conductivity of a material is quantity that characterizes the heat transfer of a material. It is the result of two contributions, the electronic conductivity and the phonon conductivity. The method tested here is used to evaluate the phonon conductivity of a material prior its implementation to the density functional theory code Demon [1].
To compute the thermal conductivity of a system from molecular dynamic simulations, several methods are possible:
- The equilibrium molecular dynamic methods, that compute the thermal conductivity at the equilibrium state base of Einstein or Green-Kubo methods.
- The non-equilibrium molecular dynamic methods, for which a gradient of temperature is imposed between to domains of the simulated box, the thermal flux is then determined before computing the thermal conductivity.
- The reverses non-equilibrium molecular dynamic methods, where a thermal flux is imposed between two domains of the simulation box, leading to a temperature gradient used to evaluate the thermal conductivity:
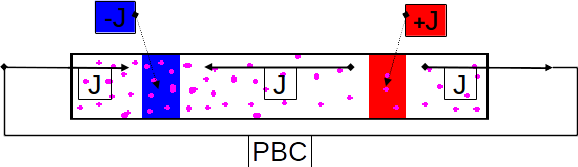
Schematic representation of the reverse non-equilibrium method.
The method was implemented and tested within a test code on solid materials, mainly rare gases considered in their solid states. A Lennard-Jones potential was used to describe the atom interactions during the molecular dynamic simulations.
Reference:
[1] Demon package: http://www.demon-software.com/public_html/program.html
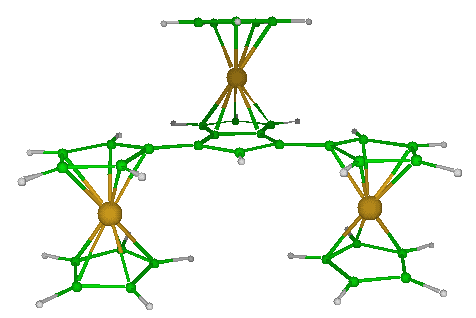 A pentalen based complex
A pentalen based complex
Licence Research Project: Versatility of Pentalen Coordination Modes in Organometallic Chemistry
2004, 3 months : Université de Rennes 1 – Laboratoire de Chimie Théorique Inorganique
Supervisors: Dr J.-Y. Saillard, Dr S. Kahlal
Coordination complexes are very important as catalysts for chemical reactions or as painting constituants due to their ability to absorb the visible light. They can vastly improve yields reducing secondary products or wastes, and thus are of economy and ecology interests. Density functional calculations involving the Amsdertam Density Functional package were employed to model pentalen complexes. The intimate relationship between the number of valence electrons and the molecular structure was investigated. Geometry optimizations and frequency calculations were carried out in order to evaluate the energies and the stabilities of an important number of molecular conformations of pentalen based complexes.
Reference:
[1] The Versatility of Pentalene Coordination to Transition Metals : A Density Functional Theory Investigation, S. Bendjaballah, S. Kahlal, K. Costuas, E. Bévillon, J.-Y. Saillard, Chemistry - A European Journal 12, 2048-2065 (2006). DOI: 10.1002/chem.200500765
2004, 3 months : Université de Rennes 1 – Laboratoire de Chimie Théorique Inorganique
Supervisors: Dr J.-Y. Saillard, Dr S. Kahlal
Coordination complexes are very important as catalysts for chemical reactions or as painting constituants due to their ability to absorb the visible light. They can vastly improve yields reducing secondary products or wastes, and thus are of economy and ecology interests. Density functional calculations involving the Amsdertam Density Functional package were employed to model pentalen complexes. The intimate relationship between the number of valence electrons and the molecular structure was investigated. Geometry optimizations and frequency calculations were carried out in order to evaluate the energies and the stabilities of an important number of molecular conformations of pentalen based complexes.
Reference:
[1] The Versatility of Pentalene Coordination to Transition Metals : A Density Functional Theory Investigation, S. Bendjaballah, S. Kahlal, K. Costuas, E. Bévillon, J.-Y. Saillard, Chemistry - A European Journal 12, 2048-2065 (2006). DOI: 10.1002/chem.200500765
