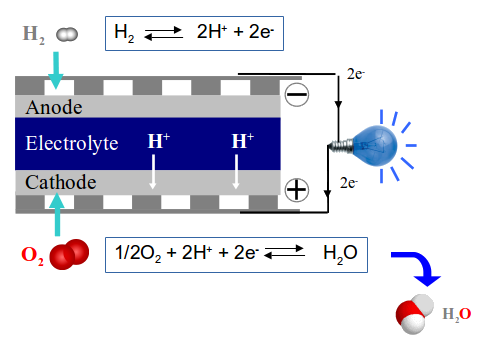
 Schematic representation of a PCFC device.
Schematic representation of a PCFC device.
Doctoral Research Project: Modeling of BaSnO3 as a Protonic Conductor for Fuel Cells
2006-2009 : École Centrale Paris – Laboratoire Structures, Propriétés et Modélisations des Solides
Supervisors: Dr G. Dezanneau, Dr G. Geneste
Protonic Ceramic Fuel Cells are electrochemical devices converting the chemistry energy of water formation reaction into electric energy. At the anode, the dihydrogen molecules are dissociated and their electrons travel within the electronic circuits contributing to the electric power. Then protons cross the dense electrolyte, joining the cathode where dioxygen molecules and electrons are waiting to terminate the chemical reaction leading to the formation of water molecules.
PCFC are a subclass of so-called Solid Oxide Fuel Cell devices, in which the moving ion is not the proton toward the cathode but the anion O2- toward the anode. The O2- anion being heavier and bigger than the proton, the activation energy required to ensure O2- displacement, and thus the migration within the electrolyte, is higher. This leads to different working temperatures, around 900°C for SOFC and around 600°C for PCFC. In both cases, the materials constituting these electrochemical devices are generally based on dense oxide ceramics, and these materials do not support frequent rise and decrease of temperatures. Accordingly their use is restricted to few start-up with long lasting operations such as auxiliary power generators. With a lower working temperature, the PCFCs induce a lower energy loss as well as a longer life for the constituting material, however this is obtained at the expense of an increasing complexity.
A general recipe to create a material suitable for a protonic conduction is to take a non-defective material, such as the stoichiometric BaSnO3 material. Then an aliovalent doping, more precisely an aliovalent substitution, is performed with an element having a lower oxidation degree. By charge compensation this potentially leads to the appearance of oxygen vacancies. In that state, the corresponding material is suitable for a SOFC device as oxygen vacancies allow oxygen displacements within the lattice. In order to obtain a PCFC, an additional hydration stage is necessary, in order for water molecules to dissociate. The oxygen of the water molecules occupy the vacancies while the hydrogen interact with the oxygen sublattice. The material is loaded of protons and can ensure a protonic conduction if the temperature is high enough. Unfortunately, the increase of temperature necessary to ensure the protonic conduction also drives a dehydration process with a decrease of the number of charge carriers. This indicates that the protonic conductivity is in fact a balance between the number of charge carriers and their conductivities, and that there is an optimal working temperature.
2006-2009 : École Centrale Paris – Laboratoire Structures, Propriétés et Modélisations des Solides
Supervisors: Dr G. Dezanneau, Dr G. Geneste
Protonic Ceramic Fuel Cells are electrochemical devices converting the chemistry energy of water formation reaction into electric energy. At the anode, the dihydrogen molecules are dissociated and their electrons travel within the electronic circuits contributing to the electric power. Then protons cross the dense electrolyte, joining the cathode where dioxygen molecules and electrons are waiting to terminate the chemical reaction leading to the formation of water molecules.
PCFC are a subclass of so-called Solid Oxide Fuel Cell devices, in which the moving ion is not the proton toward the cathode but the anion O2- toward the anode. The O2- anion being heavier and bigger than the proton, the activation energy required to ensure O2- displacement, and thus the migration within the electrolyte, is higher. This leads to different working temperatures, around 900°C for SOFC and around 600°C for PCFC. In both cases, the materials constituting these electrochemical devices are generally based on dense oxide ceramics, and these materials do not support frequent rise and decrease of temperatures. Accordingly their use is restricted to few start-up with long lasting operations such as auxiliary power generators. With a lower working temperature, the PCFCs induce a lower energy loss as well as a longer life for the constituting material, however this is obtained at the expense of an increasing complexity.
A general recipe to create a material suitable for a protonic conduction is to take a non-defective material, such as the stoichiometric BaSnO3 material. Then an aliovalent doping, more precisely an aliovalent substitution, is performed with an element having a lower oxidation degree. By charge compensation this potentially leads to the appearance of oxygen vacancies. In that state, the corresponding material is suitable for a SOFC device as oxygen vacancies allow oxygen displacements within the lattice. In order to obtain a PCFC, an additional hydration stage is necessary, in order for water molecules to dissociate. The oxygen of the water molecules occupy the vacancies while the hydrogen interact with the oxygen sublattice. The material is loaded of protons and can ensure a protonic conduction if the temperature is high enough. Unfortunately, the increase of temperature necessary to ensure the protonic conduction also drives a dehydration process with a decrease of the number of charge carriers. This indicates that the protonic conductivity is in fact a balance between the number of charge carriers and their conductivities, and that there is an optimal working temperature.
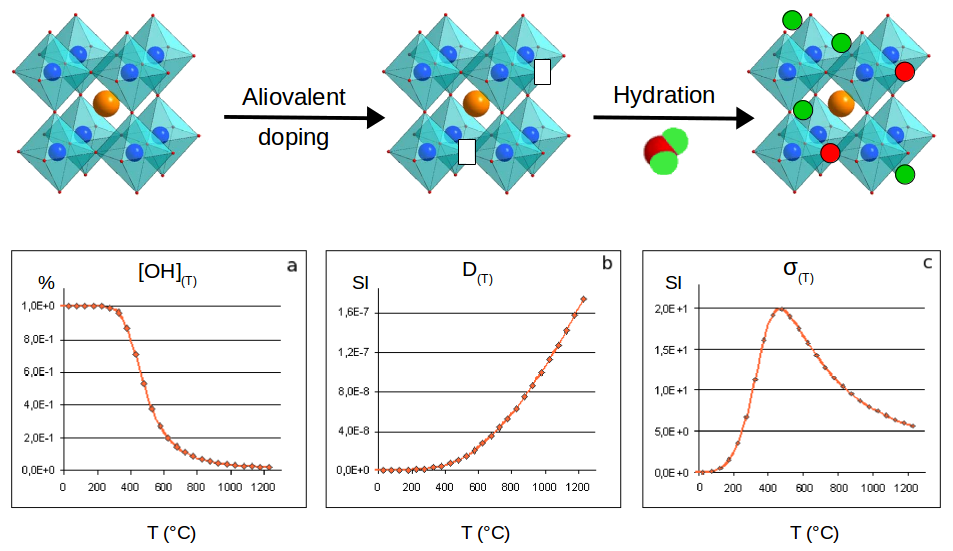
On top, schematic representation of a general method to generate a PCFC. On bottom, typical evolution of the proton concentration a), proton diffusion b) and protonic conductivity c) in a PCFC as a function of the temperature.
The situation is even more complex when one considers the possible interaction between defects, i.e. vacancy or protons on one side and substituents on the other side. To elucidate substituent effects on hydration energies in BaSnO3, we performed a systematic analysis based on the use of the Density Functional Theory as implemented in the Abinit code. Through the modeling of charged defects, we evaluated intrinsic BaSnO3 hydration energies, dopant-vacancy and dopant-proton interactions and hydration energies as a function of the defect location with respect to the substituent. The substituent investigated consisted in elements of various size, namely Ga, In, Y, Gd, Sm and La. We found a clear stabilization effect of the dopant operating on both the vacancy and the proton which was specially strong when the defect was in the direct vicinity of the dopant. These interactions increase the hydration energies compared to intrinsic material properties. This is an interesting result as it potentially increases the number of charge carriers and reduces the material dehydration while increasing the temperature in operation conditions. However, it was found that the dopants showing a size too different compared to the substituted elements led to strong lattice distortions. These distortions induce strong hydrogen bonds having the potentiality to trap the proton. This lowers the migration ability of the proton and thus reduces the protonic conductivity. For the studied concentrations of substituents, these observations led to the conclusion that the best substituents are those whose the size is the closest to the substituted element.
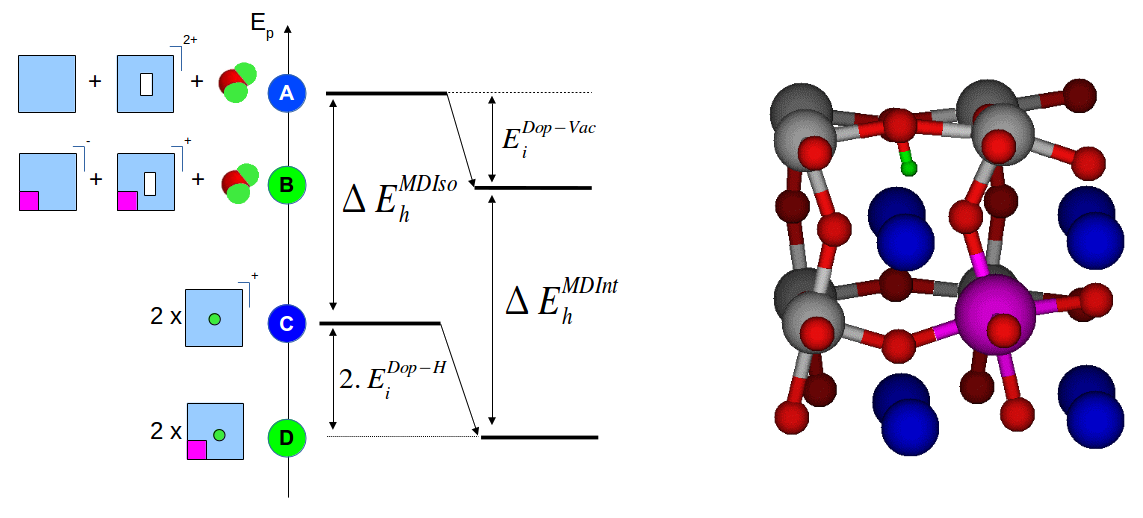
On the left, an energy diagram representing the energy levels of a BaSnO3 structures with incorporated defects. The blue squares stand for BaSnO3 cell, the white rectangles for oxygen vacancies, the purple squares represent substituants and the green dots correspond to protons. The interaction between defects lead to stabilizations and the energy difference between the vacancy state and the protonic states provide the hydration energy. On the right, illustration of a strong interaction between a proton and a substituant driving strong lattice distortions (proton in green and substituant in purple).
References:
[1] PhD manuscript: https://tel.archives-ouvertes.fr/tel-01245208/
[2] Theoretical and experimental study of the structural, dynamical and dielectric properties of perovskite BaSnO3, E. Bévillon, A. Chesnaud, Y. Wang, G. Dezanneau and G. Geneste, Journal of Physics: Condensed Matter, 20, 145217 (2007). DOI: 10.1088/0953-8984/20/14/145217
[3] Unstable polar mode and minimum of the dielectric constant in cubic BaSnO3 under hydrostatic pressure, E. Bévillon and G. Geneste, Physical Review B, 75, 214106 (2007). DOI: 10.1103/PhysRevB.75.214106
[4] Ab initio study of La-doped BaSnO3 proton conductor, E. Bévillon, G. Geneste, A. Chesnaud, Y. Wang and G. Dezanneau, Ionics, 14, 4 (2008). DOI: 10.1007/s11581-008-0218-z
[5] Hydration properties of BaSn0.875M0.125O3-δ substituted by large dopants (M = In, Y, Gd, and Sm) from first principles, E. Bévillon and G. Geneste, Physical Review B, 77, 184113 (2008). DOI: 10.1103/PhysRevB.77.184113
[6] Oxygen incorporation in acceptor-doped perovskites, E. Bévillon, G. Dezanneau and G. Geneste, Physical Review B, 83, 174101 (2011). DOI: 10.1103/PhysRevB.83.174101
[7] How dopant size influences the protonic energy landscape in BaSn1-xMxO3-x/2 (M = Ga, Sc, In, Y, Gd, La), E. Bévillon, J. Hermet, G. Dezanneau, G. Geneste, Journal of Materials Chemistry A, 2, 460-471 (2014). DOI: 10.1039/C3TA12870A
